Tutorial: Running QMrebind on the HSP90 System
In this tutorial, we will be parametrizing the HSP90 protein with an inhibitor using the LEAP and Antechamber tools. We will then replace the inhibitor partial charges using QMrebind to generate more accurate parameters (for simulation in SEEKR2 or another application).
Initial Parametrization
Let us start with the protein databank (PDB) crystal structure 2BSM (where the inhibitor is also known as compound 3)[1].
Follow the link to the PDB page for HSP90/inhibitor: https://www.rcsb.org/structure/2BSM.
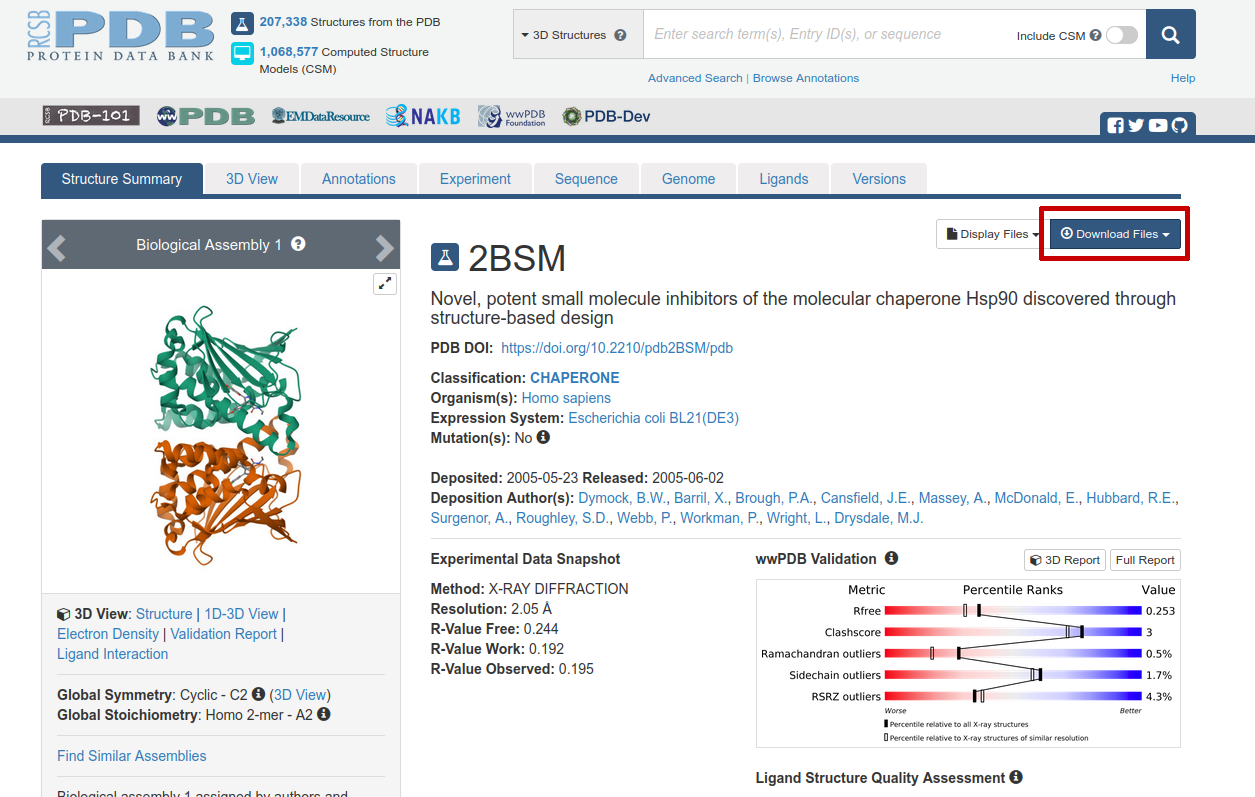
Figure 1: The Protein Data Bank entry we will use as a starting structure for our qmrebind calculations of HSP90/compound3 (ID: 2BSM).
Click on “Download Files” near the top right (circled in red in Figure 1). Then select “PDB Format”. Save the file “2bsm.pdb” somewhere on your hard drive.
Further down on the same page, one should download the CIF file for the inhibitor:
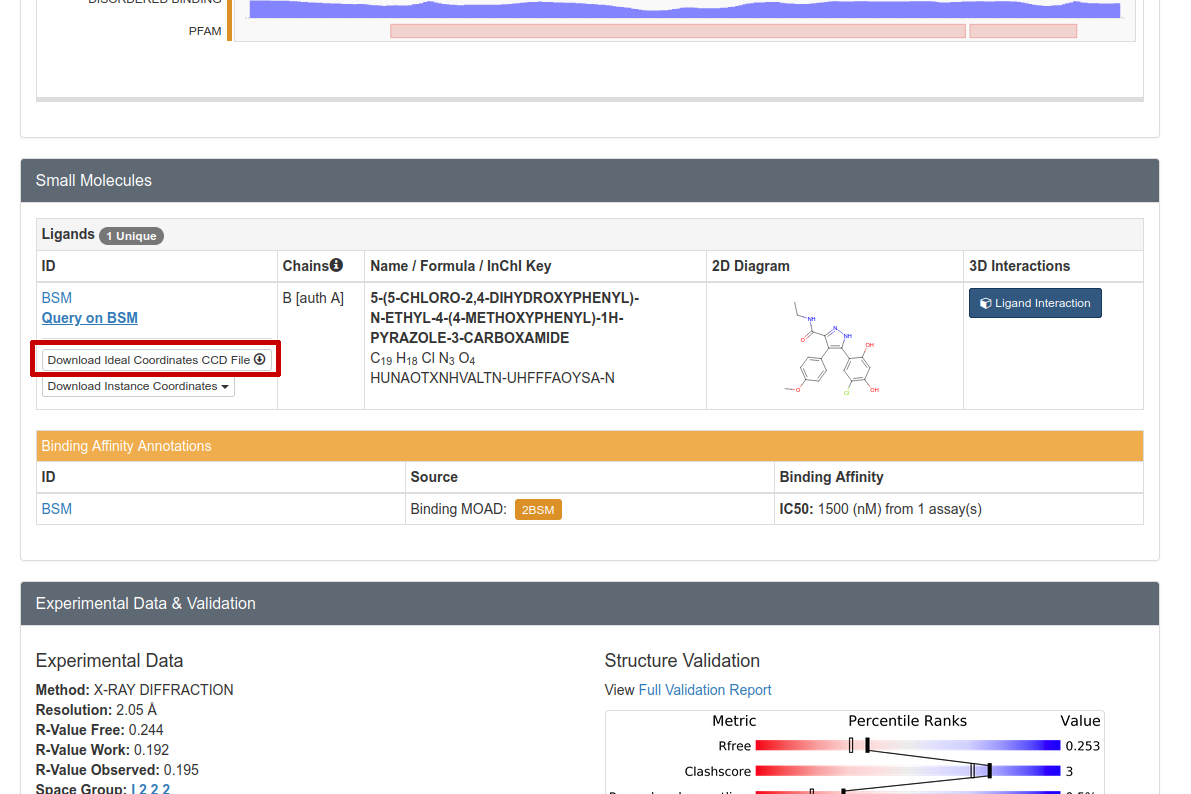
Figure 2: We must download the CIF file in order to parametrize the small molecule inhibitor.
Click on “Download Ideal Coordinates CCD File” near the top right (circled in red in Figure 2). Then save the file “BSM.cif” somewhere on your hard drive.
At this stage, many people will “clean” their PDB file using various tools. For instance, the Schrödinger commercial software, if available to you, includes a PDB cleaner.
Note
This process has been greatly simplified for demonstration purposes. When preparing your own system for a QMrebind calculation, many other procedures should be observed, including, for instance, assigning the correct protonation states to histidine residues. You should either consult an expert at preparing molecular dynamics (MD) simulations, or become an expert yourself before proceeding with your own system(s) of interest.
Use Antechamber to Parametrize Ligand
We must perform an initial parametrization of the small molecule inhibitor. One way to do this is with the Antechamber program in AmberTools.
Here, we will use Antechamber to parametrize our ligand: the small molecule “compound 3”.
Figure 3: Compound 3 inhibitor of HSP90, resname: “BSM”.
Antechamber requires a structure file to parametrize the small molecule. In this case, we will use a CIF file we just downloaded.
First, see if you have Antechamber:
which antechamber
If this returns a path, you should be good. Otherwise, you’ll need to install AmberTools. If antechamber exists on your computer, run the following command:
antechamber -i BSM.cif -fi ccif -bk BSM -o BSM.mol2 -fo mol2 -c bcc -nc 0
Each argument means the following information:
-i BSM.cif - Take the BSM.cif file as the input structure.
-fi ccif - The format of BSM.cif is in CIF format.
-bk BSM - The component/block ID for compound 3 in the PDB file is “BSM”.
-o BSM.mol2 - Specify output file name of compound 3 molecule.
-fo mol2 - Output the BSM.mol2 file in MOL2 format.
-c bcc - Use the AM1-BCC semi-empirical method to assign partial charges of the atoms.
-nc 0 - This molecule has no net molecular charge.
Use the parmchk2 program to generate a frcmod file, which LEAP will need to create the bound system:
parmchk2 -i BSM.mol2 -f mol2 -o BSM.frcmod
Then, generate a “.lib” file which will contain a library of forcefield parameters for the compound 3 molecule. Enter the following commands into the terminal:
tleap
source leaprc.gaff
BSM = loadmol2 BSM.mol2
saveoff BSM BSM.lib
quit
Use LEAP to Create the Forcefield Parameters for the Solvated System
Note
You should remove all CONECT records from your PDB files before using LEAP.
Now with a text editor, copy the following script to a file named leaprc:
source leaprc.protein.ff14SB
source leaprc.gaff
source leaprc.water.tip3p
set default PBRadii mbondi2
loadoff BSM.lib
loadamberparams BSM.frcmod
WAT = TP3
HOH = TP3
loadAmberParams frcmod.tip3p
mol = loadpdb 2bsm.pdb
solvatebox mol TIP3PBOX 10
addIons2 mol Na+ 0
addIons2 mol Na+ 24
addIons2 mol Cl- 24
saveamberparm mol hsp90_compound3.parm7 hsp90_compound3.rst7
savepdb mol hsp90_compound3.pdb
check mol
charge mol
quit
Then, run LEAP with the following command:
tleap -f leaprc
If everything runs correctly, the files hsp90_compound3.parm7 and hsp90_compound3.rst7 should be generated.
Download any Missing Files
If anything went wrong with any steps above, you can download the files below to use for the next step.
Reparametrize with Qmrebind
Now, let’s reparametrize compound 3 with more realistic partial charges. The inhibitor itself has the resname BSM, so enter the following command:
python ~/qmrebind/qmrebind/run_qmrebind_amber.py hsp90_compound3.pdb hsp90_compound3.parm7 -L BSM -Q -1
Notice that we had to specify that the QM2 region has a charge of -1 using the ‘-Q’ argument. This is because of a deprotonated aspartate close to the inhibitor in the crystal structure.
References
1. Kokh DB, Amaral M, Bomke J, Grädler U, Musil D, Buchstaller HP, Dreyer MK, Frech M, Lowinski M, Vallee F, Bianciotto M, Rak A, Wade RC. Estimation of Drug-Target Residence Times by τ-Random Acceleration Molecular Dynamics Simulations. J Chem Theory Comput. 2018 Jul 10;14(7):3859-3869. doi: 10.1021/acs.jctc.8b00230.